

Question: suggest improvements for this experiment and any areas for errors. The seperation was hard to see, suggest an alternative solvent system that will work better
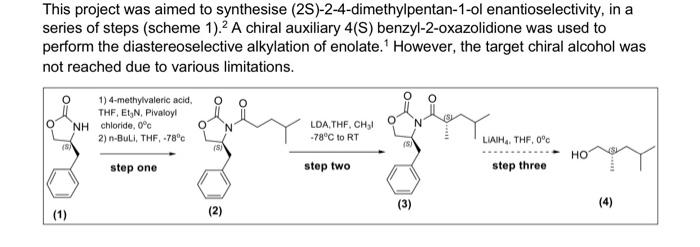

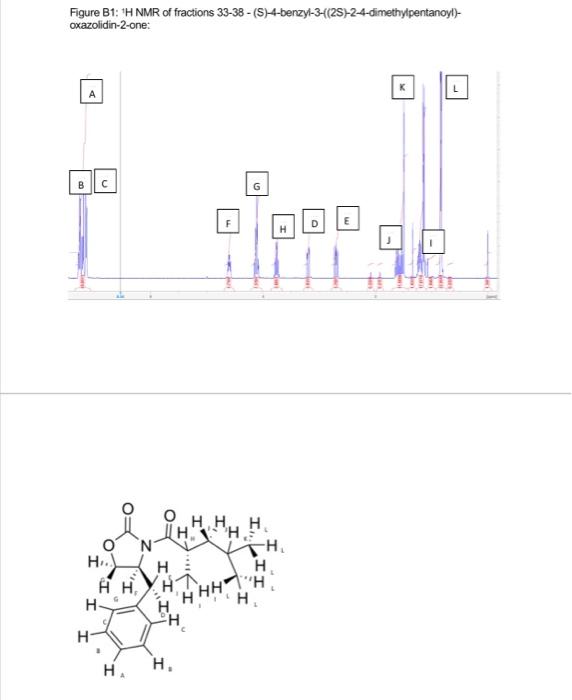
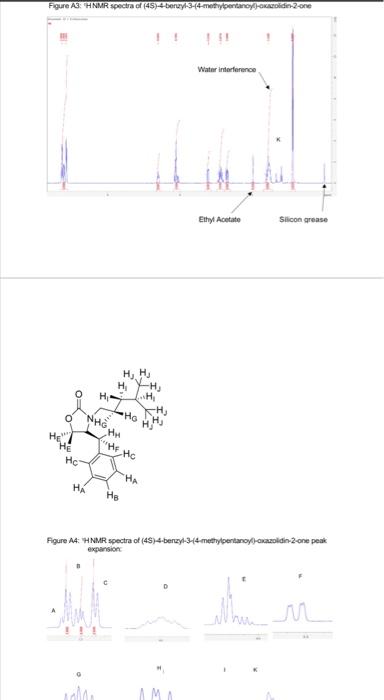
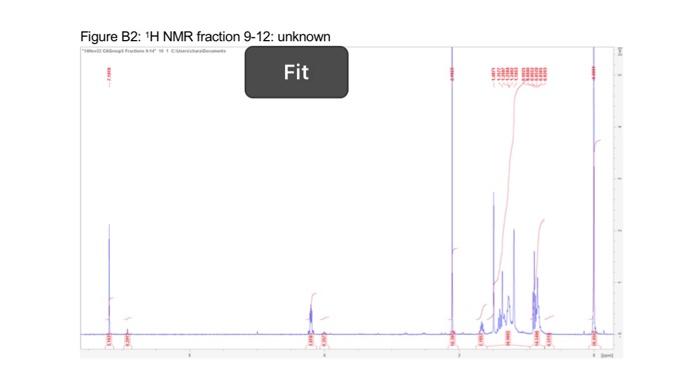
This project was aimed to synthesise (2S)-2-4-dimethylpentan-1-ol enantioselectivity, in a series of steps (scheme 1). 2 A chiral auxiliary 4(S) benzyl-2-oxazolidione was used to perform the diastereoselective alkylation of enolate. 1 However, the target chiral alcohol was not reached due to various limitations. Al experimental procedures was carried under an inert atmosphere of nitrogen. Thin layer chromatography was performed using Merck silica gel (60F-254, plated 0.25mm ). Low temperature baths used in this experimental procedure were: 78CC (dry ice and iscpropanol), 30C (dry ice and isopropanol) and 0C (ice and water). Carbon and proton magnetic resonance spectra (IIC NMR and 'HNMR) were recorded on a Bruker Avance Ill HD NanoBay 400MHz NMR spectrometer. NMR analysis was carried out on Burker TooSpin 4.1.2. Where chemical shifts were expressed in parts per million (ppm) relative to the internal relerence CDCl2 ('' C77ppm, ' H7.25ppm ). Infrared spectra was recorded and obtained on a Perkin-Elmer Spectrum two FT-IR spectrometer. Synthesis of (4S)-4-4benzyl-3-(4-methylpentanoyl) oxazolidine-2-one: 4.methylvaleric acid ( 0.40mL,2.72mmol,1.2eq) was added to anhydrous THF (15mL) and Was at 0C forming a colourless solution. Pivaloyl chorlide (0.46mL, 2.95, 1.3eq) and triethylamine (0.6mL, 3.86mm i, 1.7eq) was added after giving a thick white suspension. The mixture was stirred at 0C for a further 40 minutes. Separatoly, (4S)-4.benzyl-2coazolidinone (0.41g. 3.09mmol, 1 eq) was dissolved in antydrous THF (15mL) and the mixture was stired at 78C.2.5M n-butylithum in hexane (1.10mL,2.73mmol,1.2ec) was added dropwise. The mixture was set to stir for 30 minutes at 7BC. The lithio-(4S)-4 benzyl-2-oxazolidinone reaction was transferred via canula to the anhydride solution and was stirred for 2 hours. The mixture was warmed to room temperature, and was coenched with saturated ammonium chloride (2mL). The reside was taken up in ethyl acetale (120mL). Whilst the THF was removed under reduced pressure. The resulting residue was washed sequentially with saturated sodium carbonate ( 340mL), saturated ammonium chlonide (340mL) and brine (340mL). The colourless organic layer was dried over magnesium sultate, filtered and concentrated in a rotatory evaporator providing a crude oil. Silica gel column chromatography ( 20% ethyl acetate in hexane) was used to purify the crude of eluting the product (4S)-4-benzyl-3-(4-methylpentanoyi)-oxazolidin-2-one as a colourless ofl. (0.31g. 1.13mmol, 50%, IR (neat oll) and NMR(20mg in 700L of CDC13).R=0.26(20% ethyl acetate in hexane); 'H NMR (400 MHz, CDCl3, 5) 7.28 (m, 2H, CH(a)), 7.19 (m. 1H, CH(b)),7.12 (d, J= 6.82Hz,2H,CH(c)),5.28 (m, 1H, CH(d)),4.40(m,2H,CH(e)),4.10 (dd. J=13.36,4.02Hz,1H,CH(f)),2.82(m,2H,CH(g)),2.80(dd,J=13.36,9.60Hz,1H,CH(h) ). 1.54 (m, 3H, CH(i)), 0.88 (d, J=6.35Hz, 6H, CHO); IR ( KBr,vcm1)2985 (CH (alkene). m) 2870 (C-H (akane), m), 1761 (CaO (carbsmate), s), 1698 (C=O(amide), s), 1605 (CnC, w). 1350 (C-N, s), 1197 (C-O, s). Synthesis of compound 3, (4S)-4-benzyl-3-((2S)-2-dimethypentanoy)-oxazolidin-2-one: A stirring solution of disopropylamine (0.30mL,1.79mmol,1.60eq) in antydrous ThF (3.5 mL) was cooled to 30C.2.5Mn-butylithium in hexane (0.62mL,1.55mmol,1.30eq) was added to the stirring solution dropwise and stirred for 30 minutes. (4S)-4.bencyl-3-(4methypentanoy)-oxazolidin-2-one (0.31g.1.13mmol,1eq) in anhydrous THF (4 mL.) was cannulated into the LDA solution at 78C and stired for 60 minutes, a yellow colour was coserved. lodomethane (0.11mL,1.78mmol,5.0eq) was added at 78C. The reaction was stirred for 60 minules and was warmed to room temperature. The reaction was quenched with saturated ammonium chioride ( 5mL ). THF was removed under reduced pressure and the reacticn was taken up in ethyl acetate ( 120mL ). The organic extract was washed sequentialy with saturated sodium bicarbonate (330mL), saturated ammonium chioride ( 3 30mL) and brine (330mL). The yellow organic solution was dried over magnesium su'tate and concentrated in vacuo to provide a crude oil (0.13g). Slica gel column chromatography was carried out to purify the residus. The elution started of with 10% dethyt ether in hexane to 50% diethyf ether in hexane. Calculation of yield for step 1 : 255346312t=1.13mmol Figure B1: ' 1H NMR of fractions 33-38 - (S)-4-benzyl-3-(2S)-2-4-dimethylpentanoyl)oxazolidin-2-one: Figure A4: HNMR spectra of (45)-4.benzyl-3-4 methypentanoy)-okazoldin-2-one peak exgansion. Figure B2: ' 'H NMR fraction 9-12: unknown
Step by Step Solution
There are 3 Steps involved in it

Get step-by-step solutions from verified subject matter experts