

Question: A sample containing an alkali sulfate is dried, weighed and dissolved in dilute HCl. Barium chloride solution is added in excess to precipitate barium sulfate,
A sample containing an alkali sulfate is dried, weighed and dissolved in dilute HCl. Barium chloride solution is added in excess to precipitate barium sulfate, and the precipitate is digested in the hot solution. The precipitate is filtered through a paper filter which is then ignited and completely ashed. From the weight of the sample and weight of the precipitate, the percentage of sulfate in the sample is calculated. The precipitation reaction is the following:
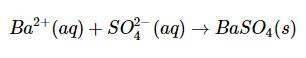
Variations in the acidity, temperature, manner of addition of the precipitant and time of digestion markedly affect the filterability of the barium sulfate precipitate and the extent to which various foreign ions are coprecipitated. Foreign anions such as nitrate, chlorate and chloride are coprecipitated as their barium salts, and the ignited precipitate contains the salt or oxide as an additive impurity. The coprecipitation of chloride can be decreased by slow addition of the precipitant. Since nitrate and chlorate interfere even at low concentrations, they should be removed from the solution before precipitation.
Foreign cations such as ferric iron, calcium and, to a lesser extent, the alkali metals are coprecipitated as the sulfates. These are substitutional impurities, and the magnitude of the error depends upon the differences between the weight of the foreign sulfate or oxide and the weight of an equivalent amount of barium sulfate. The presence of ferric iron can produce errors as high as 2% in the determination.
Preparations
Wash three 100 mm watch glasses, three stirring rods and three 400 mL beakers; rinse them thoroughly with tap water and then with distilled water. Number the beakers on the ground areas for identification using a graphite pencil. Cover each beaker with a watch glass, and store the equipment until needed.
Transfer the unknown sample to a clean dry weighing bottle. Place the uncovered weighing bottle in its upturned cap in a small beaker, cover the beaker with a dry watch glass, identify the beaker with a graphite pencil and dry the sample in a 105-110 oC oven for at least one hour.
Store the dried sample in the desiccator until it has cooled to room temperature.
You are going to transfer to three 400 mL beakers three samples weighing in the vicinity of 0.35 g but known to a precision of ±0.0001 g. You will use the method of weighing by difference.
For additional instruction on the use of balances in the laboratory and information on how to handle a weighing bottle without touching it, click here.
Weighing by difference: Take your three 400 mL beakers, your weighing bottle containing your sample and your lab notebook to the weighing room. Weigh the covered weighing bottle to a precision of ±0.0001 g. Uncover the weighing bottle and gently tap a small amount of unknown into the first beaker. Turn the weighing bottle upright again and tap it gently to recover in the weighing bottle any quantity of unknown that may still be caught on the rim. Cover the weighing bottle and weigh the contents again. Continue to do that until the difference between a subsequent weighing and your first weighing is near 0.35... g (but known to ±0.0001 g). That difference then is the weight of your first sample. Repeat the process using the other two 400 mL beakers. Care must be taken both not to lose any of your sample that stays on the rim of the weighing bottle then falls off OUTSIDE of the weighing bottle onto your laboratory bench. You must also be careful not to dump into your beaker a far greater amount than 0.35 g.
Weighing directly: The accuracy of our analytical balances is retained up to and a little beyond the mass of a 400 mL beaker. If you choose to weigh your samples directly you must be sure that your 400 mL beakers are clean, dry and at room temperature. Dry so that evaporation won't give you a negative systematic error and at room temperature so that convection currents won't give you either a positive or negative systematic error. Place the first 400 mL beaker on the balance pan and close all balance doors. Momentarily press the "tare" button and watch to make sure the balance readout shows 0.0000 g. Watch it for 10-15 seconds to make sure that it doesn't change. If it changes, press the tare button again and watch it until it is stable. Using a clean and dry spatula, transfer between 0.32 and 0.38 g of sample into the beaker. Close all balance doors and record the mass to ±0.0001 g. If you choose this method you must take care not to lose any sample on the balance pan OUTSIDE the beaker or that you don't drop any sample on the lip of the beaker from where it might fall off OUTSIDE the beaker. Repeat the process using the other two 400 mL beakers.
Add 50 mL distilled water to the sample in each beaker, then 5 mL of 6 M HCl and then add another 200 mL distilled water. Cover the beakers with the 100 mm watch glasses and store them in your cabinet until it is convenient to proceed with the determination.
Precipitation
Heat the solutions prepared above on a small hot plate to about 90 °C. Boiling the solution must be avoided since it is possible to lose some of the solution through spattering. However, it is necessary to keep the solution at an elevated temperature in order to facilitate the formation of large, filterable particles and to minimize coprecipitation of foreign ions. If, at this stage, you are using a thermometer to measure the temperature of the solution you must be very careful to rinse it with distilled water before removing it completely from the beaker. A thermometer or stirring rod when inserted into the solution will upon withdrawal remove a significant amount of solution.
Add 5% barium chloride solution dropwise from a buret which is mounted above the beaker. After 15 - 20 mL have been added, interrupt the process, allow the precipitate to settle, and test for completeness of precipitation by adding a few more drops of barium chloride. If you detect the appearance of some fine precipitate as the drop of barium chloride solution moves downward through the solution add an additional 5 mL of the barium chloride solution. Cover the beaker, and heat it on the hot plate for an hour at 90 °C. This process of "digestion" will aid in the formation of larger and purer crystals of barium sulfate. After an hour the precipitate should be coarse enough to settle readily after stirring, and the supernatant liquid should-be clear.
Filtration and Washing
Obtain three glass funnels and a wooden funnel holder from the drawers at the front of the laboratory. Clean the funnels and support them in the holder above numbered beakers or flasks of suitable volume.
The filtration will be carried out using glass or plastic funnels fitted with ashless filter paper. Ashless filter paper is pure cellulose which decomposes in the presence of heat and air to water and carbon dioxide. No residual non-volatile substances remain. Ashless filter paper comes in the form of circles which must be folded appropriately to trap all of the barium sulfate precipitate. Click here to see how to fold ashless filter paper.
In this manner insert three Whatman, ashless #42 filter papers into the funnels. Heat 200 mL of distilled water to 80C for later washings.
It is convenient to filter the barium sulfate from a hot solution since the speed of filtration is greater at the higher temperature (the solubility loss is insignificant). Care must be exercised not to lose any precipitate while transferring the filtrate (liquid) and precipitate (solid) to your filter paper.Click here for some illustrations of the experiment and proper techniques for decantation and filtering of precipitates.
Decant the supernatant through the filter, and then with the aid of the rubber policeman and small washes of hot, distilled water transfer the precipitate into the filter funnel. Since the precipitate readily clings to the side of the beaker you must carefully scrape the side of the beaker with the rubber policeman and using small water washes remove any adhering particles. After all the precipitate has been transferred wash the material in the funnel with three 5 mL portions of hot distilled water. Collect each washing separately in a small, clean beaker and then add two drops of AgNO3solution. The appearance of a cloudy, white precipitate indicates that the precipitate is still contaminated with chloride ion. If after three washings you still observe some cloudiness wash the precipitate a fourth time. Remove the filter paper from the funnel and fold it, as shown in the Web link above, and then place it in one of your crucibles. Be sure to record the identification of the sample which is stored in each crucible. If it is inconvenient to ash the filter paper immediately, store the crucibles in covered beakers in you cabinet. Do not store the crucibles in your desiccator!
Ashing and Taking to Constant Weight
Be certain that you are familiar with the points made about ashing in the Web link, above, before proceeding with the ashing step.
Support the crucible in a wire triangle and begin the heating with a small flame. Move the flame around so that all parts of the crucible are evenly heated. Gradually increase the size of the flame. Avoid heating the sample so strongly that the paper catches on fire. If it should do so quickly remove the flame and wait until burning ceases. As soon as the paper has been charred, increase the temperature of the flame. Again, move the flame as required so that all parts of the crucible get heated strongly. When all the carbon residue as been removed, the temperature should be maximized by bringing the tip of the blue cone of the flame to a point just below the wall of the crucible. Heat the crucible in this manner for ten minutes. Allow the crucible to cool for a few minutes; then place it in the desiccator and let it cool to room temperature. After weighing, repeat the process until successive heatings and weighings agree to within 0.2 mg.
Calculate the percentage of SO42- in the unknown for each sample, the average percentage and the average deviation for your results.
Report
On the report sheet, give the following information:
- The unknown number
- The weight of unknown used for each sample
- The weight of the precipitate for each sample
- The percentage of SO42- in each sample
- The average percentage of the three samples
- The average deviation from the mean of the percentage of the three samples
- Pages in your lab notebook containing the pertinent data
Questions on Sulfate Analysis
- Approximately how many mL of 5% BaCl22H2O solution would be required to precipitate all the sulfate if we assume that your samples are pure sodium sulfate? Assume that the density of the barium chloride solution is 1.00 g/mL.
- If the samples were pure potassium sulfate would you require a smaller or larger volume of barium chloride solution than the amount calculated in 1. above?
- If ordinary filter paper, instead of ashless paper were used, how would your experimental results be affected? Would they be too high or too low?
- Why are the washes of the barium sulfate tested with AgNO3?
- Does the solubility of BaSO4 increase significantly as the temperature of the solution is increased?
- What are the most important errors in this procedure?
- From you answer to question 6. above, would you say that the procedure for the sulfate analysis is likely to give results that are too high or too low?
Ba+ + (aq) + SO (aq) BaSO4(s)
Step by Step Solution
3.49 Rating (162 Votes )
There are 3 Steps involved in it

Get step-by-step solutions from verified subject matter experts