

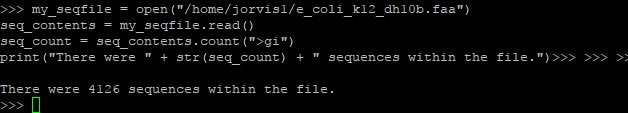
Question: In python, the following script gives the following output: my_seqfile = open(/home/jorvis1/e_coli_k12_dh10b.faa) seq_contents = my_seqfile.read() seq_count = seq_contents.count(>gi) print(There were + str(seq_count) +
In python, the following script gives the following output: my_seqfile = open("/home/jorvis1/e_coli_k12_dh10b.faa") seq_contents = my_seqfile.read() seq_count = seq_contents.count(">gi") print("There were " + str(seq_count) + " sequences within the file.")
The next question is: Make a copy of your solution from #4 and instead write a function called fasta_sequence_count which does this functionality. It should accept a file name as an argument and return the number of sequence entries found within it.
I know I need to define a function to include the functionality above, but I'm stuck.
>>> seq seq my_s eqfile = open ("/home/jorvisl/e. Coll_k12-dh10b.faa") contents my seqfile . read() count = seq contents.count (">gi") - There were 4126 sequences within the file
Step by Step Solution
There are 3 Steps involved in it

Get step-by-step solutions from verified subject matter experts